OPLS-AA topology generation is a very complex task because of huge amount of the atomtypes described this force field. The OPLS-AA force filed contains more than 800 atom types that is nevertheless not sufficient for describing all the diversity of chemical structures. Thus, as parameters for new chemical fragments appears in literature the atomtype database is also extending. TPPMKOP is an utility developed for the automatic topology generation working well with OPLS-AA force field. Below we describe the main algorithm of the atomtype attribution implemented in our TPPMKTOP project.
TPPMKTOP is free web-service available on the link: http://erg.biophys.msu.ru/tpp. TPPMKTOP works with MySQL database that contains the force field parameters and information about atomtype attribution. This database is continually upgrading by our research group. TPPMKTOP is opensource project and we can provide access to the latest mercurial version on request (comconadin at gmail.com).
Let’s describe the principle of the TPPMKTOP functioning on the example PDB-file of the following molecule:
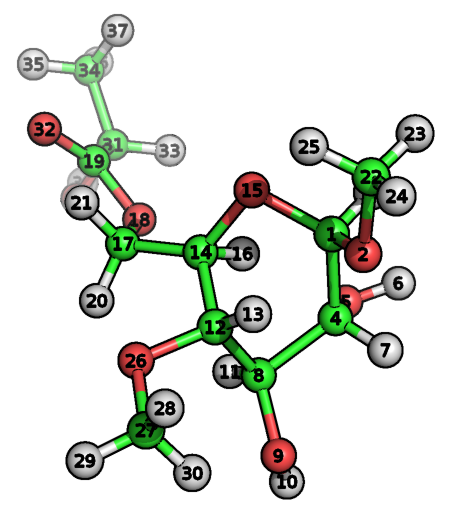 |
ATOM����� 1� C�� LIG���� 1����� -1.409�� 2.132� -0.108� 1.00� 0.00���������� C ATOM����� 2� O�� LIG���� 1����� -2.614�� 2.502� -0.908� 1.00� 0.00���������� O ATOM����� 3� H�� LIG���� 1����� -0.928�� 3.050�� 0.331� 1.00� 0.00���������� H ATOM����� 4� C�� LIG���� 1����� -2.042�� 1.200�� 1.158� 1.00� 0.00���������� C ATOM����� 5� O�� LIG���� 1����� -1.172�� 1.333�� 2.365� 1.00� 0.00���������� O ATOM����� 6� H�� LIG���� 1����� -1.313�� 2.283�� 2.719� 1.00� 0.00���������� H ATOM����� 7� H�� LIG���� 1����� -3.067�� 1.619�� 1.316� 1.00� 0.00���������� H .... |
When one uploads the PDB-file on the server the following command is executed:
tppmktop -i file.pdb -f OPLS-AA -o file.itp -r file.rtp -v -n -l lack.itp
where file.pdb is your uploaded file and OPLS-AA is the force field name. Program makes 3 output files: file.itp with standalone topology, file.rtp for making topologies by pdb2gmx, and lack.itp file with missing force field parameter definitions. All the output files, log file and console output become available once tppmktop finishes its work. Let’s consider the details of the topology preparation process for our example molecule.
(STEP) First, program prints its internal statistics.
Input file format: Protein Data Bank. Forcefield OPLS-AA was found in database. Description: Optimized Potential for Liquid Simulation. All-atomic variant.. Total statistics: 865 atoms, 314 bonds, 988 angles, 1269 dihedrals, 0 nonbonded parameters.
In the console output TPPMKTOP prints the statistics of the server database. These values correspond to the total number of atomtypes and bonded interaction parameters defined for the selected force field in the database.
(STEP) Then TPPMKTOP read and process the input chemical structure. The program writes console output:
Atoms: [.......................................]
.. and information to the log file at the same time
Trying to read structure from 'file.pdb'. LIG - C: 1(1) [ -1.409, 2.132, -0.108] QMname: C LIG - O: 2(2) [ -2.614, 2.502, -0.908] QMname: O LIG - H: 3(3) [ -0.928, 3.050, 0.331] QMname: H LIG - C: 4(4) [ -2.042, 1.200, 1.158] QMname: C ...
Here you can check if your atom names, coordinates and element names (it is important!) are attributed correctly.
(STEP) Next step is construction of the atom covalent bond matrix (showing what atoms are bonded) that is not printed in log. This step is programmed with openbabel external routines. One can check its work by converting your PDB into SMILES format.
(STEP) After the bond matrix is built the SMARTS fitting procedure is initiated. It is reflected both in console output:
3 queries proceeded on database Calculating scores for every atom.. finished! Patterns are loading. Please wait.. finished. Starting SMART-fit. Patterns checked: 296.
.. and in log, where all SMARTS patters are enumerated:
Starting curious SMART-fitting procedure. Loading patterns from database...OK! [OB] Process PAT: [H,C]C(=O)OC having 5 atoms. [OB] Process PAT: ClC(Cl)(Cl)Cl having 5 atoms. [OB] Process PAT: Cl[CX4;$(C(Cl)(Cl)(Cl)Cl)] having 2 atoms. [OB] Process PAT: [+NH4] having 1 atoms. ...
TPPMKTOP tries to match every pattern of the database to the chemical structure of the molecule and constructs an ordered list of fitted atomtypes for every atom.
(STEP) At the final stage of SMART-fit procedure chooses a single atom type for every atom basing on the system of conditional priorities:
Starting atom_alig.. Filling map.. Applying scores...
To understand the details of this procedure let’s look inside the core database. After tppmktop finishes the processing of our molecule�acetal opls_193 type will be attributed to the atom #1 in the final ITP-file. Why? In the SMARTS database there are two records which SMARTS match the chemical environment of the atom #1. The first one is (green):
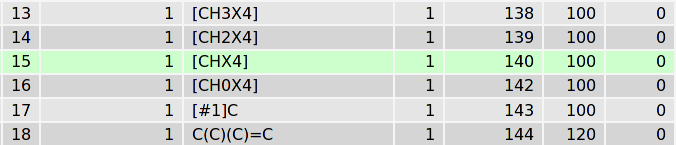
«[CHX4]» corresponds the aliphatic carbon atom with one hydrogen and exactly 4 valencies. The second pattern is:

This record means that 3rd atom of the «C-O-[CH](C)-O» pattern (bold) should be defined with atom type with ID 196. This SMARTS pattern describes the following fragment: aliphatic carbon atom bonded to oxygen that bonded to aliphatic carbon with only one hydrogen, one aliphatic carbon and one oxygen that connects two carbons. The choice between two patterns is performed according to the conditional priority («good» attribute): 196th atomtype have 150 score points whereas 140th atomtype have only 100. Thus, the 196th atomtype will be chosen. In the internal enumeration 196th atomtype corresponds to opls_193.
(STEP) On this step atoms are grouped into charge-groups according to the database and impoper dihedrals (keeping planarity or chirality) are attributed as well. The algorithm is similar to SMARTS-fitting described above.
CHARGEGROUP patterns are loading. Please wait.. finished. Starting SMART-fit. Patterns checked: 8.......... Renumbering CGNR according to human-readable style..finished. IMPROPER patterns are loading. Please wait.. finished. Starting SMART-fit. Patterns checked: 2.
(STEP) If the force field design implies explicit adding of 1-4 interaction into [pairs] section then it will be automatically performed:
Generating 1-4 pairs for FF needs..ok.
(STEP) Undescribed force field parameters are written to lack.itp file as separate #define constructions with zero coefficients. You can easily complete the topology by filling lack.itp file and copying its content to the head of the main ITP file. For our example structure there are no undescribed bonded interactions:
TPP will write 0 lack parameters to lack.itp.
(STEP) At the last stage tppmktop prints the summary charge of the system. If all atom types are attributed correctly than this charge will equal (or close) to the total charge of your molecule. If some inequality remains you can correct some partial charges manually.
Please, correct your charges according to sum: 0.000. TPPMKTOP finished normally!
Finally, you should check if the assigned atom charges are in agreement with the intuitive view on the overall chemical structure. All atomtype attribution comments are printed after semi-colons:
[ atoms ] 1 opls_193 1 LIG C 1 0.300 12.011000 ; C(HCO2): acetal OCHRO 2 opls_186 1 LIG O 2 -0.400 15.999400 ; O: acetal ether 3 opls_194 1 LIG H 1 0.100 1.008000 ; H(CHO2): acetal OCHRO 4 opls_158 1 LIG C 3 0.205 12.011000 ; all-atom C: CH, alcohols 5 opls_169 1 LIG O 4 -0.700 15.999400 ; O: diols 6 opls_170 1 LIG H 5 0.435 1.008000 ; H(O): diols 7 opls_140 1 LIG H 3 0.060 1.008000 ; alkane H. 8 opls_158 1 LIG C 6 0.205 12.011000 ; all-atom C: CH, alcohols 9 opls_169 1 LIG O 7 -0.700 15.999400 ; O: diols 10 opls_170 1 LIG H 8 0.435 1.008000 ; H(O): diols 11 opls_140 1 LIG H 6 0.060 1.008000 ; alkane H. 12 opls_183 1 LIG C 9 0.170 12.011000 ; C(HOR): i-Pr ether, allose 13 opls_185 1 LIG H 9 0.030 1.008000 ; H(COR): alpha H ether 14 opls_183 1 LIG C 10 0.170 12.011000 ; C(HOR): i-Pr ether, allose 15 opls_186 1 LIG O 11 -0.400 15.999400 ; O: acetal ether 16 opls_185 1 LIG H 10 0.030 1.008000 ; H(COR): alpha H ether 17 opls_490 1 LIG C 12 0.190 12.011000 ; C(H2OS) ethyl ester 18 opls_467 1 LIG O 13 -0.330 15.999400 ; AA -OR: ester 19 opls_465 1 LIG C 13 0.510 12.011000 ; AA C: esters - for R on C=O, use #280-#282 20 opls_469 1 LIG H 12 0.030 1.008000 ; methoxy Hs in ester</pre> ...